
�����ڵ�λ�ã� �ټ�����ҩ������ҳ >> ҩƷר�� >> �༪��|�������� >> �ٴ��о�
�༪�����ٴ�ҩ������ѧ�о�
- ��Դ�� �о����ֲ������ ���ߣ��ټö�̬ ����� ����ʱ�䣺2007/7/30 11:45:00
����
�˲�������Ŀ�����ṩ��Sorafenib�ٴ�ҩ���о��������ܽᡣ�˽����ᵽ���о�����Ϊȷ����ҩ���������еļ����������о�������ҩ��ѧ�о�����л;�����о�����������Ⱥ���о��Լ�Ϊ����Sorafenib�����������еĿ���ҩ������ʱ��ҩЧѧ����ȫ���о��������о��������2004��12��31�ձ������˽ڻ����������ձ����еļ����������о�11497���õ���ҩ������ѧ���ϣ���2005��4��22�ձ�������
����֢�����н��е�I���о�Ŀ����Ѱ���ʺϵĸ�ҩ�������Դﵽ��ĸ�ҩ�������Ӱ��ҩ��Ũ�ȣ���˶Զ��ָ�ҩ���������˹۲졣����ЩI���о��еó��İ�ȫ�Ժ�ҩ������ѧ�Լ�ҩ���Ч�������ϴ�ʹ�ں�����չ���ٴ�II���о��в���400mgÿ�����ε�������ҩ������
Sorafenib��ҩ������ѧ�����ʾ��¶���ڻ�����ڽϸ߲��죬����ϵ���ٷֱȣ�%CV����Χ��36%��91%���뵥������ȣ��ڷ�400mgÿ�����ε���̬Cmax�� AUC(0-12)��ֵ�ֱ�������4����4.7�����뵥������ȣ��ڷ�400mgÿ�����ε���̬Cmax�� AUC(0-12)��ֵ�ֱ�������4����4.7������ʳ��֬����ʳ��Sorafenib�����ս�����Լ29%����ˣ��ڷ���Sorafenibʱ��Ӧ��ʳ�е�֬����ʳ���߽�ʳ��Sorafenib�밢ù��������ҩ����ù����HCC���ߵ�AUC��ֵ������21%���в��������������Ƿ�����ٴ�����ԡ�
Sorafenib�������濵������ҩ�������濵�Ļ��Դ�л����SN-38�ı�¶���������ӣ�67%��120%����SN-38����UGT1A1���д�л����������õ��ٴ������δ����
ҩ������
Sorafenib��ҩ������ѧ�о����õķ������������о��ߵ��θ�ҩ�Ͱ�֢����ÿ�����θ�ҩ���ڽ���־Ը�����е���֬����һ�η���400mg��Sorafenib���սϿ죬��λ���Ũ��ʱ��tmaxԼ4-8Сʱ��ƽ�����Ũ��Cmax��Χ1.67-2.13mg/L��Ѫ��Ũ��-ʱ��������ʾ��1-2��tmax��������շ壬��ζ���������ѭ������̬ҩ������ѧ��֢���߷���SorafenibƬ��400mgÿ�����κ���ж��������о�100342��100277��10164��Ѫ��ҩ������ѧ�����ڵ�һ�Ƴ����һ�θ�ҩ���ռ�����Sorafenib����λ����ʱ��tmax,ssΪ3Сʱ����Χ0.0-24Сʱ����tmax�������շ�����ڸ�ҩ��8-12Сʱ��/��24Сʱ��ŷ�ͱ������е�I���о��л���ƽ����̬��Cmax,ss��AUC(0-12),SSֵ�ֱ�Ϊ7.7mg/L��64.3mg*hr/L��������5-7��
�� 5-7��������10164��100277��100283��100342�У���֢���߿ڷ���ҩ��ÿ�����Σ�ÿ�η���400mg Sorafenib��Ѫ����Sorafenib�����л��������̬Cmax�� AUC(0-12)����������ƽ��ֵ��%CV��
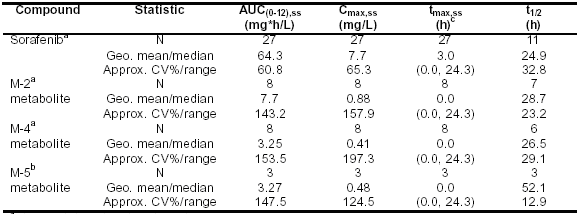
a ��ҩ����7������̬����
b ��ҩ����21������̬����
c tmax��ƽ��ֵ����Χ
t1/2 ��˥�ڣ���ѪҩŨ�ȣ�ʱ�����ߵ�ĩ��б���й�
Sorafenib��ҩ������ѧ�����ʾ��¶���ڻ�����ڽϸ߲��죬����ϵ���ٷֱȣ�%CV����Χ��36%��91%��
��������
I���ٴ����������˶������ҩ��Sorafenib��ѪҺ�е��������Ȼ��������൱��IJ��죬������Ԥ�Ƶĵ�һSorafenib����ĩ��������˥�ھ�ֵԼΪ25��48Сʱ�Ľ��������Ƚ�����10164�л���7���21��ı�¶�����ݣ����ָ�ҩ7��֮��Sorafenib�ı�¶���������ӣ����Sorafenib�ı�¶���ڸ�ҩ��7��֮�ھʹﵽ����̬���뵥������ȣ��ڷ�400mgÿ�����ε���̬Cmax�� AUC(0-12)��ֵ�ֱ�������4����4.7����
����������ö�
ͨ���뺬��PEG������-80����Һ���Աȣ�����50mg SorafenibƬ��������������öȡ�����Һ����ȣ��뵥������ȣ��ڷ�400mgÿ�����ε���̬Cmax�� AUC(0-12)��ֵ�ֱ�������4����4.7����
��ʳ��Ӱ��
Ϊ���о���ʳ��Ӱ�죬�ֱ������˽���־Ը�߷��õ�����Sorafenib��ͬʱ��ʳ��֬����ʳ���е�֬����ʳ���ʳ״̬�µ�ҩ������ѧ����ʳ�е�֬����ʳ��SorafenibƬ�����������ö����ʳ������ơ����ʳ��ȣ���ʳ��֬����ʳ��Sorafenib�����ս�����Լ29%����ˣ��ڷ���Sorafenibʱ��Ӧ��ʳ�е�֬����ʳ���߽�ʳ��
ÿ��һ����ÿ������
�ֱ�������Sorafenib�����ַ�ʽ��ҩ��ҩ������ѧ��������200mg��QD����100mgÿ�����Σ�bid�����ڷ���100mg bid�Ļ��ߣ�24Сʱ��AUCֵ��200mg QD���߸�������ÿ�����θ�ҩ���������ö����ӣ����ܹ�����Sorafenib��ˮ�еĵ��ܽ�ȣ�����θ�������β��ֵ����ռ��١���Ϊ��ÿ��һ������£����Ÿ�ҩ���������ӣ��������öȴﵽ���Ե���̬ˮƽ����������ѡ��ÿ�����εĸ�ҩ��ʽ�Դﵽ������ջ�������ճ̶ȡ�
����������ϵ
�ڵ���400mg bid������AUC�������߶�������ͨ��������й������������������ϵ��������600mg bid����AUC(0-12),ss��ֵ��400mg bid��Ȳ�δ�ɱ������ӣ���800mg bid����AUC(0-12),ss��ֵ��600mg bid���Ҳδ�ɱ������ӡ�
�ֲ�
99.5%��Sorafenib��Ѫ������ϣ���ϳ̶���Ũ�ȳ�������ء�Sorafenib�ں�Ѫ���Ѫ���еķֲ�������ȣ�Ѫ����ѪҺ֮��Ϊ1.33��
����
Ϊ�о�Sorafenib����й�ʹ�л;��������11195��������λ����־Ը�߽�������������ƽ�����顣�ڷ�������[14C] Sorafenib������Լ�ⷢ��Sorafenib��Ҫ�������й��Լռ�������������77%����Һ��ռ19%��ԭҩ�Ƿ����ȡ���е���Ҫ�ɷ֣���ҩ������50.7%���������M-6�����ữ�����ռ��ҩ������19%������Һ��й��19%�У���Ҫ�ɷ֣�14.8%����Sorafenib�������ᣨM-7������2.7%��M-2�������ᡪM-8����Һ��δ��Sorafenibԭҩ��Ѫ��������������У�73%��Sorafenib��ʽ����ѪҺѭ����17%Ϊ��л����M��2������ҩ��ѧ�������ơ�Ѫ��������������Դ�л���������1%��
���Դ�л�����ҩ������ѧ
I �ں�II���ٴ������о���Sorafenib�Ĵ�л����M-2��M-4��M-5�ڰ�֢�������ڵ�ҩ������ѧ��ͼ5��1������Щ���ݱ�����400mg bid�������������ҩ��M-2�ı�¶��Լռ�ܱ�¶����ԭҩ�ʹ�л�����ܺͣ���16%����л����M-2��M-4�ڸ�ҩ��7��ﵽ��̬����̬ʱ��л����M-4ռ�ܱ�¶����8%��M-5ռ6%���Ƚϴ�л����M-5�ڸ�ҩ���1��͵�21���AUC��Cmaxֵ�����ֵ�21��M-5��¶�������Ӹ���Sorafenib��M-2��ͼ5��1����
���ڴ�л
�����Sorafenib�ڴ���(73)��Ȯ(74)����(63)���ڵ�����ת�����飬������4��5��ʾ����ĿǰΪֹ���ڷ�������ҩ���ԭҩ�Ǵ���Ȯ����Ѫ���е���Ҫ��֣��ڷ������AUC������ռ71%��73%����������ȣ������ȮѪ����M��3��BAY 72-1973�������Ƶ�λ���ֱ�ռ�������AUC������12.1%��15.6%������Ѫ����M��2��BAY 67-3472, N-�����������Ҫ�Ĵ�л���AUC������16.7%���������ڴ���Ѫ���м�����M��2��0.9%������ȮѪ������δ�����
��4��5����������ת�����ڷ�������[14C]��ǵ�BAY54��9085����(PH-33292)��Ȯ(PH-33063)����(PH-33427)Ѫ���е���Ҫ���

a ��[14C]Sorafenib�����ɼ����
AUC ѪҩŨ��ʱ�����������
�����壬Sorafenib��ѪҩŨ��ʱ�����߳��ּ����μ��壬�������ڸγ�ѭ�����γ�ѭ���ij��ֹ������������ֹ��̣�
�� ҩ����������ữ����M��7�����ܷ��ڵ����������������ֽ⣬���Sorafenib�ڳ��������ա�
�� ҩ�����������M��2�����ܷ��ڵ��������᳦����ԭΪSorafenib�����Sorafenib�ڳ��������ա�
��������۲쵽������Ҫ������ת��;�����ڴ����Ȯ��δ���ָγ�ѭ���������Ȯ�ڷ�[14C]��ǵ�Sorafenib�ױ������κ�����Լ���ȫ���������й��������Լ75%�ĸ�ҩ�����������й��ԭҩ���ˣ���ҩ������50.7%���ʹ���(32.3%)�����ȡ���е���Ҫ��֣�����Ȯ��ռ��ҩ������3.5%�����ˣ������Ȯ�ķ����ȡ���У����ữ����M��6�ֱ�ռ�������������19.1%��30.0%��62.6%���Ǽ�������M��3(BAY 72-1973)�����Ƕ������ڶ��еĴ�л����ڴ����Ȯ�ķ���зֱ�Ϊ23.0%��15.0%��
�����壬���������й�ȴ����Ȯ��Ϊ���ԡ����˵���Һ��������19%���м����ִ�л���M��7��Sorafenib���������ữ���������14.8%����M��8��M��2���������ữ���������2.7%��������δ����ԭҩ�ɷ֡��Ƚ�Sorafenib������Ͷ��������ת�����֣�������л;������������ڴ����������졣�����壬��व�N��������Ӧ(M-2, BAY 67-3472)�ȼ��ǻ�����Ӧ(M-3, BAY 72-1973)��Ϊ������Sorafenib���������ữ���������ھ�����Ҫ���塣
����������������Sorafenib������;����������ת������CYP3A4����M��2�������Լ���������ȩ��ת��øUGT1A9�����������ữ����M��7�����ɡ����⣬��������Ҳ�������������ữ����M��7�����ڴ�����������ת��;����һ����ΪSorafenib��CYP3A4���Ƽ���̫���ܷ���ҩ�������á����ٴ������У�CYP3A4��ǿЧ���Ƽ�ͪ������BAY54��9085������ҩ����Sorafenib��ѪҩŨ�Ȳ�δ�����κ�����Ӱ�죬����ܵ�ԭ�����������ữ��л;������������(75)��
ͼ 5-1���ڸ�ҩ21��/ͣҩ7������飨����10164���У���֢���߷���600 mg bid������Sorafenib���1��7��21�죬Sorafenib�����л����M-2��M-4��M-5��ѪҩŨ��
 ����������л����M-5�İ�˥�ڽϳ�һ�¡��ձ���֢�����ڵ�14��͵�28������ݱ�������ҩ14����л����M-5�ﵽ��̬������Sorafenib�������ִ�л���M-2��M-4��M-5�������Ƶ�����ҩ��ѧ���ԣ����Ҹ�ռ��Ѫ��������ҩ���й�����AUC������30%���ң���ô������ѪҺѭ����Sorafenibû����Ҫ�Ļ��Դ�л���
����������л����M-5�İ�˥�ڽϳ�һ�¡��ձ���֢�����ڵ�14��͵�28������ݱ�������ҩ14����л����M-5�ﵽ��̬������Sorafenib�������ִ�л���M-2��M-4��M-5�������Ƶ�����ҩ��ѧ���ԣ����Ҹ�ռ��Ѫ��������ҩ���й�����AUC������30%���ң���ô������ѪҺѭ����Sorafenibû����Ҫ�Ļ��Դ�л���������ʱ��������
����������
��I �ڵ�һ�Ƽ������У�������Ѫ���е�Sorafenib�����л����M-2��M-4��M-5�����ż�������ߣ������������൱����Sorafenib�����������������ʣ�Sorafenib��M-2��M-4��M-5����������ռ�ı���δ�����Ա仯��������100mg bid��800mg bid������Χ�ڣ�Sorafenib�Ĵ�л���м��������ԡ�
ʱ��������
�ڼ����ٴ��������о���Sorafenib��ҩ������ѧ��ʱ��������ԣ�����������10164�������˸�ҩ���1��7��21���ȫ��ѪҩŨ��-ʱ�����ߡ���Щ���������Ŀ���ǿ���������ҩʱ���Sorafenib�����л�����ҩ������ѧ��Ӱ�졣���鷢�ֶ������ҩ����7���Sorafenib�� Cmax��AUCֵ�����ټ������ӡ�����10164�У��Ƚϸ�ҩ���7�����21��ı�¶�������ֶ������ҩ��Sorafenib��M-2��M-4��AUCֵ���������ӡ�
��л����M-5���γɽ�������˲����ܴӵ�1�����������M-5��Ũ�ȡ��Ӹ�ҩ���1�쵽��7��M-5�ı�¶�����ӣ���ҩ��7�쵽21��������ӣ����Ǹ�ҩ��21�쵽28��ı�¶�������˵��M-5�ı�¶���ڶ������ҩ��21���ڴ���̬ˮƽ�����ձ����е�����10658�У������˸�ҩ���1��14��28���ҩ������ѧ������ÿ�ո�ҩ���κ��14��28��M-5��Cmax�� AUC(0-12)ֵ���������10658������10164�����ݱ�������ҩ���7��10��M-5�ı�¶���ﵽ��̬����л����M-5���γɽ��������˥�ڽϳ�����ʵ�����
��֮������10164��100277��100283��100342��10658�����ݱ�����Sorafenib��M-2��M-4��M-5�ڸ�ҩ��14���ھ��ﵽҩ������ѧ��̬ˮƽ��
�������ص�Ӱ��
������Sorafenib��ҩ������ѧ���������صĹ�ϵ���������ذ������䡢�Ա����ء�Ѫ�弡�����Լ�������ʼƣ����ι��ܣ�Child��Pugh A����Child��Pugh B�����Լ�Ѫ�嵨���صȣ���һ�ع�������ʹ����I �ں�II ���ٴ�������Sorafenib�����黼�ߵ����ݣ���Щ����õ��˻���������ѪҩŨ��-ʱ�����ݡ�Sorafenib��ҩ������ѧ�����䡢�Ա����ء�Ѫ�弡����Ѫ�嵨����û�����ԵĹ�ϵ����Child��Pugh A����B�����ߣ�Sorafenib��ϵͳ��¶���Ͱ�ȫ���������ơ��Ƚ�Sorafenib���ձ��������ߵ�ҩ������ѧ������Sorafenib�ڲ�ͬ���ߵı�¶��������һ���̶����ص���
���ϣ������ѵõ���400mg bid���ݣ�����������䡢�Ա����أ���Χ��51��116���������ҩ������
ҩ���ҩ������ѧ�����
Sorafenib��ϸ�������Կ���ҩ����ڰ�֢����ͨ����ͬʱʹ�ö���ҩ��������ƣ����Ժ��б�Ҫ�о��ٴ���Sorafenib������ҩ���ҩ������ѧ����á�ͨ�������л/����/�յ���P-gpת�˺���������������ҩ���DZ�ڵ���������ԡ����⣬�ٴ���������ijЩҩ��������á�����ҩ�ᆳ������������ҩ����ʹ�ã���������������Sorafenib����������ҩ���������ҩ�������ṩ������ɵ��������ݡ�
�������������������������Sorafenib�Ĵ�л�ص㡣Sorafenib�������л����ȷ����I �ں�II �������еĴ�л;�������������о���Sorafenib��9����CYP�칹���5����UDP-����ȩ��ת��ø���������á��������ϸ�������������о���Sorafenib��ø�յ����á���Ϊ�������۵�һ���֣����ٴ�����ƽ������Ͱ�֢����I ��������Ҳ�о���Sorafenib�Ĵ�л����֢�����ٴ������п�����Sorafenib��CYP�칹��ѡ���Ե����Ӱ�졣�����ּ�Ҫ�ܽ���������������ڴ�л����Ľ����
1����������
�����˸�������������ʾ�� Sorafenib������л;��I��Ҫ��CYP3A4��������л;��II�������������ϣ���Ҫ��UGT1A9������л����M-1��M-5��������л���M-6���ữ��л���M-7��M-8�ֱ���Sorafenib��M-2���������ữ���
��������������Sorafenib�ױ������ζ�9����CYP�칹����������á�δ��Sorafenib��CYP1A2��CYP2A6��CYP2E1���Բ����������ã�Sorafenib��CYP2C19��CYP2D6��CYP3A4�����ж��������ã�Kiֵ�ֱ�Ϊ17 M��22 M��29 M��CYP2B6��CYP2C8��CYP2C9�Ļ����ܵ�Sorafenibǿ���������ƣ�Kiֵ�ֱ�Ϊ6 M��1��2 M��7��8 M��Sorafenib�Դ�л;��II����Ϸ�Ӧ��øUGT1A1��UGT1A9Ҳ�����������ã�KiֵΪ1��2 M����400mg bid������Sorafenib����̬ѪҩŨ��Cmax��ֵΪ16.6 M��7.7 mg/L��������Sorafenib��Cmaxֵ�������ױ����Ķ���Kiֵ�����Sorafenib���ܶ���ЩCYPs��������ڴ�л����DZ���������á�
�ٴ���������������ܵ�Sorafenib���Ƶ�CYPͨ�����ٴ�������ԡ���ΪSorafenibͨ������ƽ�е�;�����д�л������Sorafenib�뵥һ;������ҩ��������ҩʱ����¶���������ӵĿ����Խ�С��ͪ�����ٴ�����������֤ʵ����һ���裬����δ�����Ե�����á�ͬ����Ҳû�з���CYP1A2��CYP3A4���յ����ã�˵���ٴ��Ϸ���ø�յ��Ŀ����Ժ�С��������10926�Ľ��һ�¡�
2������P-�ǵ��ף�P-gp����������
Caco-2ϸ����ͨ�����۱���Sorafenib�Ǹ��������Caco-2ϸ������P-gp��Sorafenib��Caco-2ϸ���ײ��������ٴӶ����ײ�ת�˵�������Ϊ2.9��4.7������Sorafenib�и����ԣ�Ԥ��������������Ӱ������������������ա�
����������ʾSorafenib�ױ������ζ�����P-gp�����þ����൱����������á�����̽�뻯�������߶�����˫�״�Ī������IC50ֵ�ֱ�Ϊ0.84µM��1.24µM�����ٴ������У���400 mg bid������Sorafenib��ѪҩŨ�ȸ߳�����IC50ֵ����֮�࣬����Sorafenib���ܶԾ�P-gp;��ת�˵�ҩ�����DZ���������á�˫�״�Ī��Sorafenib(102)һ����P-gp�����������á�����˫�״�Ī��������ظ�����¶���������ӣ����Sorafenib��P-gp����������ҩʱ���ٴ��Ͽ��ܲ��ᷢ�����Ե�����á���˽�����һ�µ����б���ָ����P-gp����ҩ�������йص��������˵���ܲ�̫��Ҫ����103��
3��CYP3A���Ƽ�ͪ�����Sorafenibҩ������ѧ��Ӱ��
�������鷢��Sorafenib��������л;��I��Ҫ����CYP3A4�������ںܶ����������ͨ��CYP3A4���д�л������ҩ���������Ҳ��CYP3A4�йأ��������ǿ�����CYP3A4��ǿ�����Ƽ�ͪ�����Sorafenib��Ӱ�졣�������У�Sorafenib��ҩ����Ϊ50 mg��ͬʱÿ�����ͪ����400 mg����7�졣�������ͪ����δ����Sorafenib�ı�¶��������ѪҩŨ�ȣ�����5��8����
�� 5-8��Ѫ����Sorafenib�� AUC��Cmax�������Լ���Щ��������ͪ��������/δ��ͪ��������ʱ�ı�ֵ(������Сƽ����ֵ) (���� 10927)
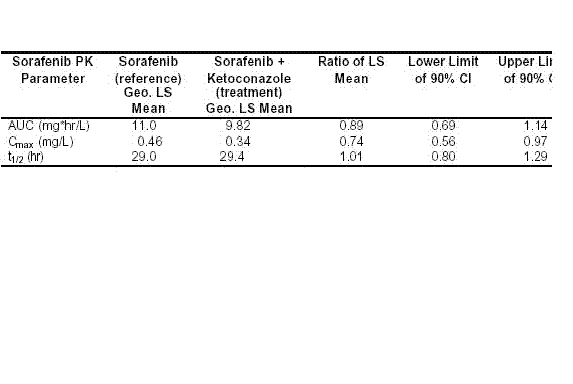 AUC = ѪҩŨ�ȣ�ʱ�����������
AUC = ѪҩŨ�ȣ�ʱ�����������CI = ��������
Cmax = ���ѪҩŨ��
PK �C ҩ������ѧ
Geo. LS mean = ������Сƽ����ֵ
t1/2 = ��˥�ڣ���ѪҩŨ�ȣ�ʱ������ĩ��б���йء�
��ͪ����������ҩʱ��Sorafenib�ı�¶��δ�����ӣ���˿����ƶϾ���CYP3A4����������л;��I��Sorafenib�Ĵ�л�в�̫��Ҫ����Щ���ݽ��III�����飨����11213�����ݣ���������41λSorafenib�����黼����19λ��ο���黼��ͬʱ����CYP3A4���Ƽ�����������ٴ���Sorafenib��CYP3A4��������Ƽ�������ҩ�ǰ�ȫ�ġ�
ͪ������P-gpת�˵�ǿ�����Ƽ��������ٴ�������δ��ͪ�����Sorafenib��ҩ������ѧ�����κ�Ӱ�죬���Կ��ƶ�P-gpת�˶���Ѫ����Sorafenib��ҩ������ѧ��˵������Ҫ��
����������ҩ�����õ����������
δ��Sorafenib�뼪���������ɳ��������һ�µĻ��������ҩ������ѧ����ã�����Sorafenib�����뼪���������ɳ����������ҩ��
Sorafenib�밢ù��������ҩ����ù����HCC���ߵ�AUC��ֵ������21%���в��������������Ƿ�����ٴ�����ԡ�
Sorafenib�������濵������ҩ�������濵�Ļ��Դ�л����SN-38�ı�¶���������ӣ�67%��120%����SN-38����UGT1A1���д�л����������õ��ٴ������δ����
4����������ϸ��ɫ��P450��Sorafenib����������
���ڰ�֢���߽��е��ٴ����飨����10926���У����߳��ڷ���Sorafenib�����쵥һCYP�칹�����������Ƿ�����ٴ�����ԣ�������Sorafenib ��CYP2C19(��������)�� CYP2D6(����ɳ��)��CYP3A4 (�������)̽������Ӱ�졣
��֢���߶�η���400 mg bid������Sorafenib��CYP3A4(�������)��CYP2D6 ������ɳ�ң���CYP2C19(��������)̽������ҩ������ѧ��δ���������仯������ٴ���Sorafenib�ȷ�CYP2C19��CYP2D6��CYP3A4ͬ��ø�����Ƽ���Ҳ�����ǵ��յ�����Sorafenib�����������ӻ���پ�����Щ;����л�Ļ�����ı�¶����
����������Sorafenib������CYP2C9��KiֵΪ7��8µM������S-��������Ҫͨ��CYP2C9;����л��������11213�У�ͨ��PT-INR������Ӽ��Sorafenib�Ի������л��Ӱ�졣PT-INR������Sorafenib���밲ο����δ�����죬˵��������Sorafenib���ܲ���CYP2C9�����Ƽ���
TAG���༪��
���ҩƷ





��վ��Ѷ- ҩ������ |
�����ŵ�ֲ� | �˲���Ƹ |
��ϵ���� | ��վ��ͼ
�������- �������� | ����ָ�� | ҩѧ���� | �˿���� | �˿�Ͷ�� | ר�Ʒ��� | Ѱҽ��ҩ | ҩʦ����
ר�Ʒ������- ������ | �β��� | �� | ����� | Ƥ���Բ��� | �� �� | ��ʪ���߿� | ��Ѫ�ܿ� | ���� | ��������ҩ
ҩƷ����- ������ҩƷ | �����ҩƷ | �β���ҩƷ | �ۿ�ҩƷ |Ƥ���Բ���ҩƷ | ��ҩƷ | ��ʪ���߿�ҩƷ
�ټ�ҩ������- ��ҵ����Ӫҵִ�� | ������ҩƷ��Ϣ�����ʸ�֤
�������- �������� | ����ָ�� | ҩѧ���� | �˿���� | �˿�Ͷ�� | ר�Ʒ��� | Ѱҽ��ҩ | ҩʦ����
ר�Ʒ������- ������ | �β��� | �� | ����� | Ƥ���Բ��� | �� �� | ��ʪ���߿� | ��Ѫ�ܿ� | ���� | ��������ҩ
ҩƷ����- ������ҩƷ | �����ҩƷ | �β���ҩƷ | �ۿ�ҩƷ |Ƥ���Բ���ҩƷ | ��ҩƷ | ��ʪ���߿�ҩƷ
�ټ�ҩ������- ��ҵ����Ӫҵִ�� | ������ҩƷ��Ϣ�����ʸ�֤
������ҩƷ�����ʸ�֤|������ҩƷ��Ϣ�����ʸ�֤��|��ICP�����ţ���ICP��17064671��-3
Copyright@2013-2021 �ټ���������ҩ��.�ټ�����ҩ���� www.xinyao.com.cn ��Ȩ���У�����������Ȩ����
�ͻ��������ߣ�400-101-6868 Ͷ���뽨�飺020-29859339
Copyright@2013-2021 �ټ���������ҩ��.�ټ�����ҩ���� www.xinyao.com.cn ��Ȩ���У�����������Ȩ����
�ͻ��������ߣ�400-101-6868 Ͷ���뽨�飺020-29859339

