
您现在的位置: 百济新特药房网首页 >> 药品专题 >> 多吉美|索拉非尼 >> 临床研究
多吉美临床前动物药效学研究
- 来源: 研究者手册第六版 作者:百济动态 浏览: 发布时间:2007/7/30 11:33:00
简要介绍
Sorafenib最初是在对CRAF激酶抑制剂先导物进行结构-活性评价的生化分析中被发现的。在体外,sorafenib是CRAF和野生型及突变型(V600E)BRAF的有效的抑制剂。Sorafenib还抑制参与肿瘤进展和血管生成的几种RTKs,包括人VEGFR-2,小鼠VEGFR-2,VEGFR-3和PDGFR-β,FLT3和c-KIT。在人乳腺癌、黑色素瘤、胰腺癌和结肠癌细胞系中,sorafenib降低了细胞内MAPK途径的基础磷酸化水平,但对表达突变KRAS的NSCLC细胞系NCI-H460或A549细胞无此作用。在表达野生或突变型KRAS或BRAF的人乳腺癌、胰腺癌、黑色素瘤和结肠癌细胞系中,sorafenib抑制了细胞内MAPK途径的信号传导。
要点:
・Sorafenib是RAF激酶的强抑制剂,所抑制的RAF激酶包括CRAF、野生型和突变型(V600E)BRAF及在肿瘤进展中发挥作用的几种RTKs。
・ Sorafenib在体内具有广谱的抗肿瘤活性。在表达突变RAS和/或BRAF的负瘤小鼠模型,及经RAS/RAF/MEK途径传导生长信号的负瘤小鼠模型中,sorafenib的治疗抑制了肿瘤的生长。
・Sorafenib通过抑制VEGFR2,VEGFR3和PDGFR-β,而减少了血管生成信号的传导。因而,可通过直接和间接机制共同抑制肿瘤生长。
体外研究
表4-1总结了sorafenib的生化活性及细胞药理学结果(21)。sorafenib,是CARF和野生及突变(V600E)BRAF的强抑制剂,体外IC 50分别为2nM,22nM和38nM(22)。sorafenib还抑制几种参与肿瘤演化和血管生成的RTKs,包括FLT3, c-KIT,VEGFR-2,VEGFR-3和PDGFR-β。即使药物浓度高达10μM,sorafenib对MEK-1, ERK-1, EGFR, HER2/NEU, c-MET, PKA, PKB, IGFR-1, Cdk-1/cyclin B, PIM-1, GSK 3-, CK-2, PKC-, PKC-ß, 或 PKC-。
Sorafenib对牛p59 FYN激酶的IC50为780nM,说明药物对此激酶没有显著的抑制作用。对非激酶靶位,sorafenib对腺苷A3,多巴胺D1和毒蕈碱M3受体有轻微的的抑制作用,IC50分别为1.6μM,2.0μM和3.1μM。
表4-1:Sorafenib对激酶靶位的体外活性
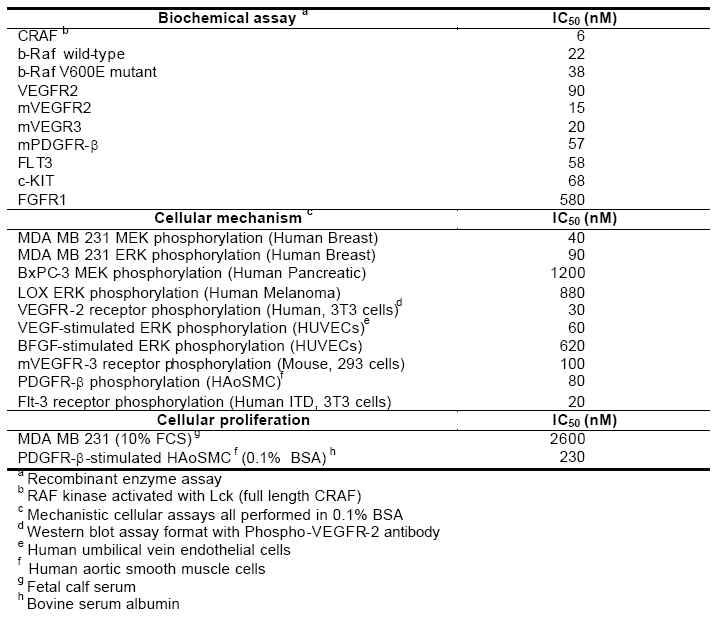
在体外,sorafenib对乳腺癌、胰腺癌、黑色素瘤和结肠癌细胞的RAF/MEK/ERK途径的信号传导有显著的抑制作用,表现为表达野生型或突变型KRAS或BRAF的细胞的pERK水平降低。然而,对于NSCLC细胞系A549和H460细胞,即使sorafenib的浓度高达20M,仍然没有观察到药物对RAF/MEK/ERK途径的信号传导有抑制作用。药物对这些细胞系内ERK磷酸化无抑制作用的原因尚不清楚。Sorafenib对表达在增生的血管内皮细胞内的(VEGFR2)、淋巴生成内皮细胞内的(VEGFR3)、和人大动脉平滑肌细胞的(PDGFR-β)靶受体的磷酸化也有抑制作用。VEGF和PDGF受体在肿瘤新血管生成中起重要作用(15, 20)。因此,sorafenib一方面通过抑制VEGFs和PDGF受体而阻断肿瘤新血管生成,另一方面通过抑制RAF/MEK/ERK途径的信号传导而抑制肿瘤生长。
体内研究
.1).单剂药效
在表达突变的KRAS(HCT-116, DLD-1, Mia-PaCa-2)或BRAF(Colo-205,HT-29)的肿瘤模型中,sorafenib显示了抗肿瘤作用。对于表达上述两种突变基因的MDA-MB-231模型,药物仍然有效。对表达野生型RAS和BRAF、但不过表达EGF和HER2生长因子受体的人卵巢癌细胞,sorafenib也显现了疗效。这些受体也是通过RAF/MEK/ERK途径传导信号。Sorafenib在这些肿瘤模型上显现出疗效,提示RAF激酶抑制剂不仅对表达RAS和/或突变的b-Raf的人肿瘤有效,对其他通过此信号传导通路传递信号的、生长因子受体过度表达的的肿瘤也有效。最后,sorafenib对表达活化的突变型FLT-3的MV4:11AML模型也有效。表4-2总结了Sorafenib在人肿瘤异体移植物模型中显示的体内广泛的抗肿瘤活性。
表4-2:在临床前异种移植物模型中,sorafenib的抗肿瘤活性
剂量从7.5mg/kg/天到90mg/kg/天时,sorafenib在RENCA小鼠RCC模型中也显示了很强的抗肿瘤活性(肿瘤生长抑制率为30%到84%)。
在无胸腺小鼠A498和CAKI-1人肾癌异种移植模型中,进行了药物的初步的抗肿瘤疗效研究。在剂量高达60mg/kg时,sorafenib仍然没有显著抑制CAKI-1肿瘤的生长(25)。然而,当sorafenib的剂量为60mg/kg时,在研究期间,70%的A498肿瘤的的大小没有加倍(26)。相对于对照组肿瘤的生长程度,本研究中药物没有显现出明显的肿瘤生长抑制作用。如果剂量提高或者给药时间延长,sorafenib可能会表现出明显的肿瘤生长抑制作用。在两种肾癌模型中,癌细胞对Sorafenib的敏感性不同,药物作用也有差别,但是原因尚不清楚。
在DLD-1结肠癌模型,多疗程sorafenib治疗是有效的(27)。单疗程Sorafenib治疗,药物对小肿瘤或大肿瘤的生长都有抑制作用。药物对肿瘤生长的延迟作用与治疗持续时间成正比,而与治疗初期肿瘤的大小无关。与单一疗程的疗效相比,两疗程、每疗程10天的治疗使肿瘤生长延迟近乎2倍,而30天的持续治疗使肿瘤生长延迟近乎3倍。值得注意的是,在第一疗程治疗中肿瘤生长受抑制,但是中止治疗后肿瘤生长也恢复了,这时开始sorafenib第二疗程的治疗仍然可以达到抑制肿瘤生长的目的。
.2).作用机制
在多种在体肿瘤模型中研究了sorafenib的作用机理,包括HT-29, DLD-1, HCT-116, 和 Colo-205 结肠癌模型, MDA-MB-231乳癌模癌模型(21, 28)。在每个研究中,负荷大约200毫克肿瘤的动物,给予型和Mia-PaCa-2胰腺sorafenib(30-60mg/kg/剂),为期5天。末次给药后3小时取下肿瘤,用Western染色和/或免疫组化(IHC)方法,评价sorafenib对RAF/MEK/ERK信号传导通路的作用。在一些实验中,肿瘤样品还进行了抗CD31的山羊多克隆抗体染色和影像分析微血管密度以评价药物对新生血管生成的抑制作用。从HT-29和Colo-205肿瘤模型得到的结果验证了这些评价得出的结论。
每组4只小鼠移植了人结肠肿瘤,当肿瘤长至至100到200毫克时,小鼠口服剂量为30mg/kg或60mg/kg的sorafenib,每日1次,共5天。用抗-pERK和抗-ERK抗体进行Western blot分析,结果表明,sorafenib抑制了HT-29内RAF/MEK/ERK信号转导通路的活化,但是对Colo-205人结肠癌无效。虽然在HT-29和Colo-205结肠癌肿瘤异种移植模型中,sorafenib都显著抑制了肿瘤的生长,但是用Western blotting或IHC的方法分别分析Colo-205肿瘤,都未能发现药物对ERK1/2磷酸化的显著的抑制作用。提示sorafenib的治疗未能有效阻断RAF/MEK/ERK途径的信号传导。在Colo-205和HT-29细胞内都存在同样的V600E b-Raf突变,大概是这些细胞异常增殖的原因。可能存在另外一条途径,而不是RAF/MEK/ERK级联反应,活化了Colo-205肿瘤内的ERK1/2。与此相反,尽管在Colo-205肿瘤样本中,sorafenib的治疗未使RAF/MEK/ERK途径的信号传导受到抑制,但是Colo-205肿瘤样本与HT-29肿瘤样本一样,微血管区面积和密度都降低了50%到80%。
代谢物的药理学
合成了sorafenib的三种代谢物(M-2, M-4和M-5),并研究了它们对野生型c-Raf和b-Raf,mPDGFR-β, mVEGFR-2和人FLT-3的生化活性(38)。M-2和M-5对mPDGFR-β的抑制作用明显强于sorafenib。然而,sorafenib对c-Raf和野生型及突变型b-Raf的抑制作用显著强于M-2和M-5。Sorafenib对FLT-3的抑制作用明显强于M-5或M-4。
总的来说,代谢物的细胞内效应与sorafenib的基本一致。ERK磷酸化水平,用于衡量RAF/MEK/ERK途径信号传导抑制程度;在MDA-MB-231乳腺癌细胞内,这些化合物对ERK磷酸化的抑制作用没有差别,IC50为90到151nM。两个代谢物,M-2和M-5,对PDGF介导的人大动脉平滑肌细胞增生的抑制作用显著强于sorafenib。NIH-3T3纤维细胞转染小鼠VEGFR2后,观察药物对VEGFR2自动磷酸化抑制作用的IC50,代谢物的IC50为12到30nM,与sorafenib相比没有显著差别。
BAY 67-3472(代谢物M-2)的抗肿瘤活性
在两个独立的研究中,用MDA-MB-231人乳腺癌NCr裸鼠模型,比较了BAY67-3472(M-2)及其母体化合物Sorafenib的抗肿瘤效应(39)。在这两个试验中,每个制剂的口服剂量为30, 60和120mg/kg,给药方法为QD×9。如前所述,采用本研究中的给药方案,sorafenib口服剂量为30mg/kg时,可完全抑制MDA-MB-231肿瘤的生长。因此,采用这一剂量直接比较两个化合物的抗肿瘤活性。此外,基于以前的经验,即当剂量超过120mg/kg时,sorafenib的全身药物浓度不再升高,因此在两个研究中BAY67-3742采用的最高剂量为60和120mg/kg。BAY67-3742 120mg/kg的剂量,已经达到本研究所使用的赋形剂PEG400/MSA溶解的最大极限。
Sorafenib组动物耐受性良好,体重仅降低4%-5%,没有动物死亡。如前所述,在此模型上Sorafenib的疗效显著,与模型对照组相比,肿瘤生长抑制率(TGI)为80%-91%。BAY67-3472的耐受性也较好,动物体重减轻了2%-7%,没有动物死亡。BAY67-3742剂量为30mg/kg时,TGIs为38%;剂量为120mg/kg时,TGIs为68%。因此,BAY67-3472的作用强度和有效性都低于sorafenib。两药给药途径及给药方案相同的条件下,BAY67-3472的剂量为120mg/kg时产生的肿瘤生长抑制作用(TGI为63%到68%),弱于sorafenib剂量为30mg/kg时产生的效应(TGI为80%到91%)。
在单独的研究中测定了NCr小鼠口服BAY67-3472后,BAY67-3472和sorafenib的药代动力学参数。BAY67-3472疗效较低与NCr小鼠口服BAY67-3472后得到的血药浓度较低有关。BAY67-3472口服剂量为30mg/kg时,得到的药物血浆达峰浓度(Cmax)近似于4mg/L, 药时曲线下面积(AUC)为25mg*h/L;而同样剂量的sorafenib得到的Cmax比BAY67-3472高10倍,为33mg/L,AUC为326 mg*h/L(40)。至少BAY67-3472的一部分抗肿瘤疗效是通过它在体内转化生成母体药物sorafenib而实现的。口服剂量为30mg/kg 的BAY67-3472后,血浆sorafenib的浓度非常低,但服药后7h后与BAY67-3472的血药浓度相同。Sorafenib口服后,再次服用BAY67-3472,得到的BAY67-3472的AUC(0-24h)约为58%-71%。口服30或60mg/kg BAY67-3472得到的各化合物的血药浓度都低于剂量比例。
总之,这些数据说明,口服sorafenib的主要代谢物BAY67-3472,有利于药物的抗肿瘤效应。在剂量比较实验中,BAY67-3472的疗效较母体化合物低。口服后得到的血浆母体药物浓度较低的事实,已经足以说明此代谢物效应较弱的原因。然而,既然sorafenib主要代谢为BAY67-3472,此研究尚不能完全阐明BAY67-3472对sorafenib抗肿瘤活性的实际贡献。
讨论和结论
基于sorafenib体外效应,即在细胞机制研究中对CRAF的抑制作用,及其体内的广谱抗瘤作用,sorafenib被选择为临床用药候选之一。
Sorafenib时c-Raf和突变(V600E) BRAF的强抑制剂,还抑制在肿瘤演化过程中起重要作用的几种RTKs,包括VEGFR-2,VEGFR-3,PDGFR-β,c-KIT和FLT-3。在表达突变RAS和/或BRAF肿瘤模型,和其他生长因子通过ras/raf/MEK途径传递信号的肿瘤模型中,用sorafenib治疗肿瘤负荷小鼠可使肿瘤生长受到抑制。除抑制肿瘤生长的作用外,sorafenib通过抑制肿瘤新血管生成,由此通过直接和间接机制控制肿瘤生长。研究了药物在人体的主要代谢物(M-2)的体内抗癌效应,结果表明代谢物的有效性和作用强度小于sorafenib。
TAG:多吉美
相关药品





网站资讯- 药房介绍 |
连锁门店分布 | 人才招聘 |
联系我们 | 网站地图
便民帮助- 常见问题 | 服务指南 | 药学服务 | 顾客意见 | 顾客投诉 | 专科服务 | 寻医问药 | 药师窗口
专科分类服务- 肿瘤科 | 肝病科 | 神经科 | 精神科 | 皮肤性病科 | 眼 科 | 风湿免疫科 | 心血管科 | 糖尿病科 | 其他科用药
药品服务- 肿瘤科药品 | 精神科药品 | 肝病科药品 | 眼科药品 |皮肤性病科药品 | 神经科药品 | 风湿免疫科药品
百济药房资质- 企业法人营业执照 | 互联网药品信息服务资格证
便民帮助- 常见问题 | 服务指南 | 药学服务 | 顾客意见 | 顾客投诉 | 专科服务 | 寻医问药 | 药师窗口
专科分类服务- 肿瘤科 | 肝病科 | 神经科 | 精神科 | 皮肤性病科 | 眼 科 | 风湿免疫科 | 心血管科 | 糖尿病科 | 其他科用药
药品服务- 肿瘤科药品 | 精神科药品 | 肝病科药品 | 眼科药品 |皮肤性病科药品 | 神经科药品 | 风湿免疫科药品
百济药房资质- 企业法人营业执照 | 互联网药品信息服务资格证
互联网药品交易资格证|互联网药品信息服务资格证书|粤ICP备案号:粤ICP备17064671号-3
Copyright@2013-2021 百济新特网上药店.百济新特药房网 www.xinyao.com.cn 版权所有,并保留所有权利。
客户服务热线:400-101-6868 投诉与建议:020-29859339
Copyright@2013-2021 百济新特网上药店.百济新特药房网 www.xinyao.com.cn 版权所有,并保留所有权利。
客户服务热线:400-101-6868 投诉与建议:020-29859339

